

Indications and steps for performing delayed sequence intubation
Posts filed under Critical care
Proning for ARDS
A review of proning in patients with ARDS
Posterior Reversible Encephalopathy Syndrome (PRES)
Brief overview of posterior reversible encephalopathy syndrome (PRES) and how it may present in the Emergency Department. This blog has been written and peer-reviewed by emergency physicians.
Accidental Tracheostomy Decannulation
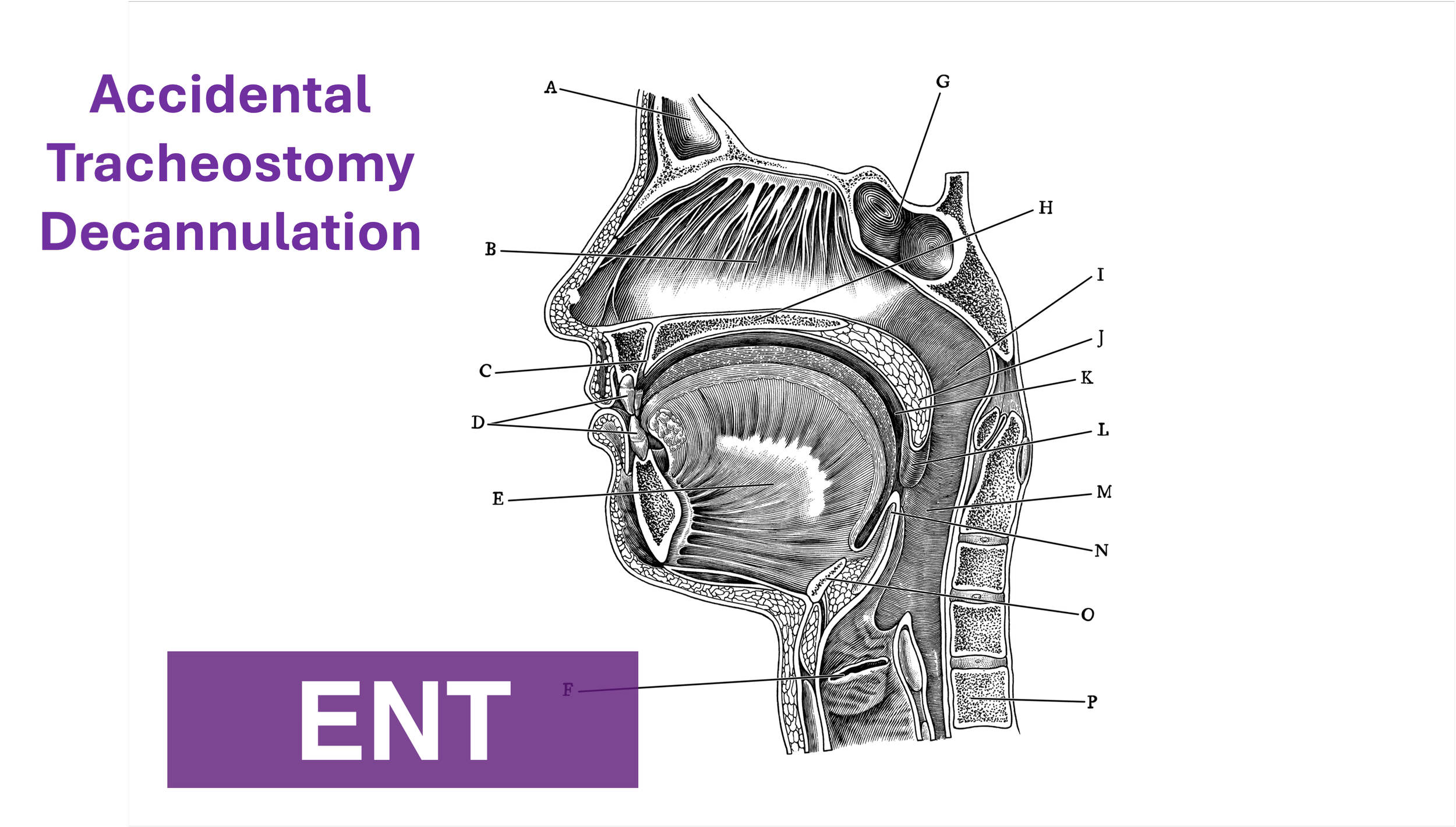
Written by: Chezlyn Patton, MD (NUEM ‘27) Edited by: Keara Kilbane, MD (NUEM ‘25)
Expert Commentary by: Matt McCauley, MD (NUEM ‘21)
Introduction
Tracheostomy is a common procedure in the US with over 110,000 trachs placed annually (1). Complications occur at a rate of approximately 40-50%, however most complications are minor, with only 1% being catastrophic (1). Of these devastating complications, 90% occur within the first 10 days of placement. Overall approximately 15% of tracheostomies will be decannulated accidentally, and in a critical care setting, 50% of airway related deaths were associated with accidental tracheostomy decannulation (1, 2).
One way to approach a decannulated tracheostomy tube could be with the acronym, IRMI; Investigate, Recannulate, Monitor, and Intubate (if you must).
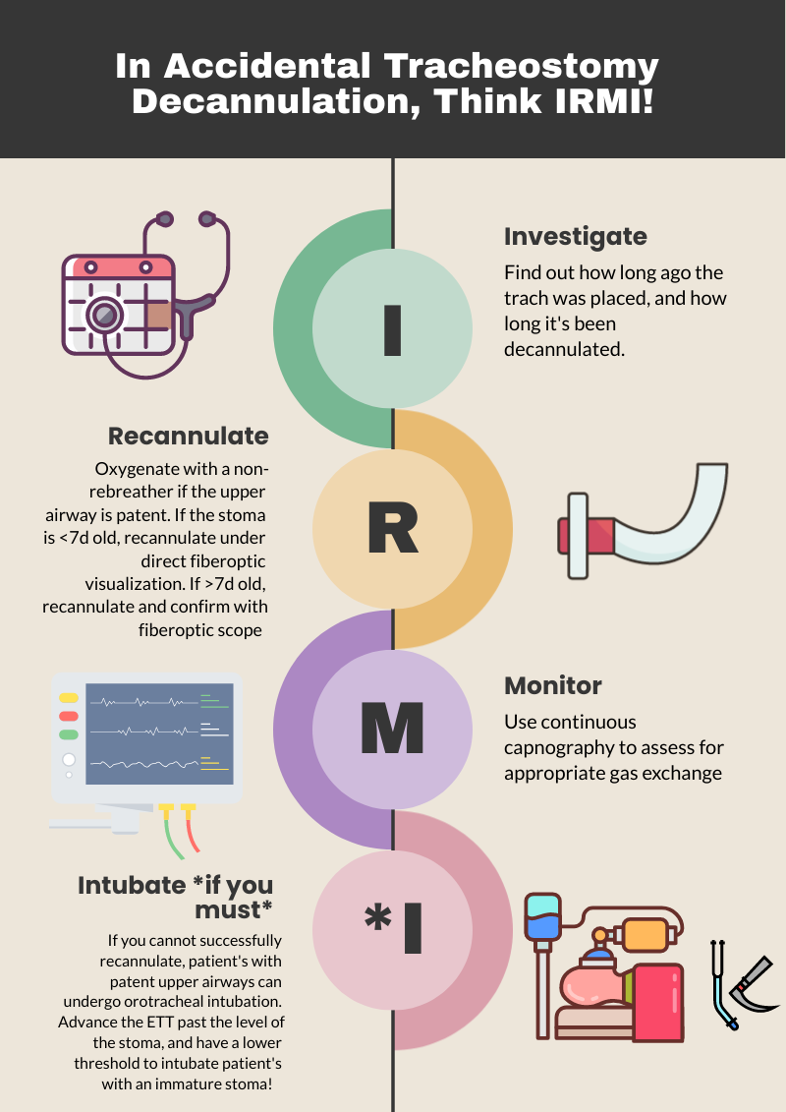
Investigate: How long ago was the tracheostomy tube placed? How long has it been out? What is the size of the trach? Is it cuffed or uncuffed? Why was the trach placed initially?
Cuffed vs. uncuffed: Is there a pilot balloon present? If yes, that indicates the trach is a CUFFED tracheostomy tube.
Size of tracheostomy tube: ALWAYS labeled on the neck flange.

Figure 1: Size of trach tube identification on flange. Borrowed from https://tracheostomyeducation.com/tracheostomy-tubes/.

Figure 2: Tracheostomy tube parts labeled, borrowed from ©Linda L. Morris and M. Sherif Afifi, https://www.trachresource.com/table-of-contents/
Recannulate: Oxygenate from above with non-rebreather or by blow by oxygen over the tracheostomy stoma if the patient is spontaneously breathing. If they are in respiratory distress or not spontaneously breathing, bag-valve-mask (BVM) oro-nasopharyngeal or over the stoma. You can use a pediatric mask to fit over the stoma or a LMA. Ensure to occlude the stoma if BVM from above or close the mouth if BVM from the stoma to prevent air leakage. Note, if the patient is ventilator dependent, you need a CUFFED tracheostomy tube. Obtain one tracheostomy tube of appropriate size, and a tube that’s a size down, as a stoma may begin to close the longer it’s out. For stomas less than 10 days old, grab a fiberoptic scope, as these will require recannulization under direct visualization. This is to minimize the risk of creating a false passage.

Figure 3: Depiction of creation of false passage through subcutaneous tissue when replacing tracheostomy tube. Borrowed from: Morris, L.L., Whitmer, A., & McIntosh, E. (2013). Tracheostomy care and complications in the intensive care unit. Critical care nurse, 33 5, 18-30 .
Otherwise, you can place blindly for the initial insertion. Ensure the obturator is placed inside the outer cannula tracheostomy tube prior to insertion, as it blunts the hard edge of the tracheostomy tube that can damage the membranous wall of trachea. If you meet any resistance, size down immediately, as the stoma has likely started to heal (even with a matured tracheostomy). Then remove the obturator and inflate the cuff to maintain placement (3,4).
Monitor: Once the trach tube is reinserted, it is important to monitor for appropriate placement and gas exchange. Continuous capnography is the gold standard for this. Additionally, the tube should be confirmed to be in the trachea through direct fiberoptic visualization of the trachea and carina. Be sure to assess for complications such as creation of a false lumen, which could manifest as subcutaneous emphysema (5).
Intubate if you must: If faced with a scenario where the tracheostomy tube cannot be passed through the stoma, and your patient is developing respiratory distress, you can intubate your patient orotracheally if they have a patent upper airway. The only exception to this is a patient who has had a laryngectomy, as those patients cannot be intubated orally and are obligate neck stoma breathers (6).
References
1.Bontempo, Laura J., and Sara L. Manning. "Tracheostomy emergencies." Emergency Medicine Clinics 37.1 (2019): 109-119.
2. Cheung, Nora Ham-Ting and Lena M. Napolitano. “Tracheostomy: Epidemiology, Indications, Timing, Technique, and Outcomes.” Respiratory Care 59 (2014): 895 - 919.
3. Rajendram, R., and N. McGuire. "Repositioning a displaced tracheostomy tube with an Aintree intubation catheter mounted on a fibre-optic bronchoscope." BJA: British Journal of Anaesthesia 97.4 (2006): 576-579.
4. Shah RK, Lander L, Berry JG, et al. Tracheotomy outcomes and complications: a national perspective. Laryngoscope 2012;122(1):25–9
5. Riley, Christine M.. “Continuous Capnography in Pediatric Intensive Care.” Critical care nursing clinics of North America 29 2 (2017): 251-258 .
6. McGrath B, Bates L, Atkinson D, et al, National Tracheostomy Safety Project. Multidisciplinary guidelines for the management of tracheostomy and laryngectomy airway emergencies. Anaesthesia 2012;67(9):1025–41.
Expert Commentary
Thank you for this concise summary of tracheostomy management. While most of the immediate complications of tracheostomy will occur in the ICU, these patients still frequent our emergency department with and without tracheostomy related emergencies. Despite this, patients with tracheostomy can be intimidating there is a general lack of knowledge about tracheostomy among healthcare professionals in general and emergency medicine trainees in specificity.1,2
As you have outlined, understanding both the chronicity of the tracheostomy as well as the indication for the procedure are key history when managing a displaced tube. A patient who underwent tracheostomy for failure to liberate for the ventilator likely has a patent upper airway while one placed following an ENT surgery likely poses a significant challenge for orotracheal intubation! Obtaining this history (and handing off this key information when transitioning care) can be lifesaving. Significant care should be taken when replacing a tracheostomy through an immature stoma, the usual cited maturity date being 10 to 14 days old. These should always be replaced over a fiber-optic scope with visualization of the tracheal rings and carina prior to cuff inflation and ventilation by BVM or ventilator. Failure to do so can result in severe pneumomediastinum, pneumothorax, subcutaneous emphysema, and respiratory arrest if placement into a false tract is not recognized3 . Replacement of a tracheostomy tube into a mature tract can be done blindly with an obturator in place but care should also be taken if there are any signs of trauma or bleeding. When in doubt, play it safe and obtain fiber optic visualization!
While trach replacement can be stressful under the wrong circumstances, patients with tracheostomy tubes can present with enumerable other emergencies. As with any high stress situation in resuscitation, it helps to fall back onto our ABCs, airway being of principle importance here. The immediate assessment of any patient with a tracheostomy tube in extremis should be focused on a singular question: can I ventilate the patient through this tube? In order for a patient to be effectively bagged or ventilated through a tracheostomy tube three things must be true. The tube must be patent and endotracheally placed, the tube must be cuffed, and the cuff must be inflated. Patency can be quickly assessed with passage of a flexible suction catheter. If this is unsuccessful, removal of the inner cannula (if present) and replacement with a fresh inner cannula can often resolve obstruction by secretions. If obstruction is unable to resolve, you should oxygenate from above while preparing to replace the tracheostomy tube as you have elegantly outlined.
The presence of a cuffed tube will be indicated by the presence of a pilot balloon, no reading of numbers or brand names needed! Finally, cuff inflation can be confirmed by palpation of the pilot balloon and assessing for any speech production or gurgling hear though the mouth. If the patient can phonate then the balloon is not properly inflated! If gentle inflation of the cuff does not resolve the air leak assume a ruptured cuff and replace the tracheostomy.
Tracheostomy tube care and emergencies can be very intimidating but this procedure is a valuable tool for ICU and ventilator liberation. As emergency physicians, we need to be familiar with the nuances of these devices so we can safely manage the airway just as we would any sick patient.
References
1. Whitcroft KL, Moss B, Mcrae A. ENT and airways in the emergency department: national survey of junior doctors’ knowledge and skills. J Laryngol Otol. 2016;130(2):183-189. doi:10.1017/S0022215115003102
2. Darr A, Dhanji K, Doshi J. Tracheostomy and laryngectomy survey: do front-line emergency staff appreciate the difference? J Laryngol Otol. 2012;126(6):605-608; quiz 608. doi:10.1017/S0022215112000618
3. Long B, Koyfman A. Resuscitating the tracheostomy patient in the ED. Am J Emerg Med. 2016;34(6):1148-1155. doi:10.1016/j.ajem.2016.03.049

Matt McCauley, MD
Assistant Professor, Division of Critical Care
UW BerbeeWalsh Department of Emergency Medicine
Associate Medical Director
UW Organ and Tissue Donation
How To Cite This Post:
[Peer-Reviewed, Web Publication] Patton, C. Kilbane, K. (2024, Apr 15). Accidental Tracheostomy Decannulation. [NUEM Blog. Expert Commentary by McCauley, M]. Retrieved from http://www.nuemblog.com/blog/tracheostomy-decannulation
Other Posts You May Enjoy







Crashing Patient on a Ventilator
Written by: Patrick King, MD (NUEM ‘23) Edited by: Adesuwa Akehtuamhen, MD (NUEM ‘21)
Expert Commentary by: Matt McCauley, MD (NUEM ‘21)

Expert Commentary
Thank you for this succinct summary of an incredibly important topic. We as emergency physicians spend a lot of time thinking about peri-intubation physiology but the challenges do not end once the plastic is through the cords. The frequency with which our ventilated patients stay with us in the ED has been increasing for years and will likely continue to do so1. This means that managing both acute decompensation and refractory hypoxemia needs to be in our wheelhouse.
The crashing patient on the ventilator can be truly frightening and your post effectively outlines a classic cognitive forcing strategy for managing these emergencies. A truism in resuscitation is to always rule out the easily correctable causes immediately. In this case, it means removing the complexity of the ventilator and making things as idiot-proof as possible. Once you’ve ruled out the life threats like pneumothorax, tube displacement, and vent malfunction, you can try to bring their sats up by bagging. Just make sure that you have an appropriately adjusted PEEP valve attached to your BVM for your ARDS patients; the patient who was just requiring a PEEP of 15 isn’t going to improve with you bagging away with a PEEP of 5.
Once you’ve gotten the sats up and the patient back on the vent, your ventilator display can provide you with further data as to why your patient decompensated. Does the flow waveform fail to reach zero suggesting breath stacking and a need for a prolonged expiratory time? Is the measured respiratory rate much higher than your set rate with multiple breaths in a row indicating double-triggering? The measured tidal volume might fall short of your set tidal volume. This points towards a circuit leak, cuff leak, or broncho-pleural fistula. Maybe you’re seeing the pressure wave dip below zero mid-inspiration and the patient is telling you that they are in need of faster flow, a bigger breath, or deeper sedation. In these situations, your respiratory therapist is going to be your best friend in managing this patient-ventilator interactions2.
As your post alludes to, sometimes patients remain hypoxemic despite our usual efforts and refractory hypoxemia can be an intimidating beast when you’ve got a busy ED burning down around you. If your cursory efforts to maintain vent synchrony by playing with the ventilator dials have failed, there’s no shame in deepening sedation which will work to decrease oxygen consumption and prevent derecruitment. Once sedated, work with your RT to find appropriate PEEP and tidal volumes to meet your goals.
Most patients can be managed with usual lung-protective ventilation but some patients will require more support and you’ve correctly identified several salvage therapies. My general approach is to pursue prone positioning in any patient with a P:F ratio approaching 150 despite optimal vent settings as it has the only strong mortality benefit of the therapies outlined above. Proning in the ED is resource intensive and is probably better pursued as a department-wide protocol rather than you and your charge nurse trying to figure it out in the middle of the night3.
As you’ve pointed out, the neuromuscular blockade has more limited evidence and is not required for prone ventilation. Upstairs, we accomplish this with continuous infusions but in the ED you may be more comfortable using intermittent boluses of intubation dose rocuronium. Just make sure your patient is unarousable. I reach for this if I’m unable to achieve ventilator synchrony with sedation alone as it allows for very low tidal volumes and inverse ratio ventilation. I see inhaled pulmonary vasodilators in a similar light: there’s no data on patient-oriented outcomes but they can make your numbers look prettier while you wait for more definitive interventions such as transfer.
This finally brings me to VV ECMO for refractory hypoxemia. It’s worth considering that while there is some evidence for a mortality benefit for ECMO in ARDS, the evidence base is mixed. The CESAR trial did show a mortality benefit in patients transferred to an ECMO center but only 76% of patients actually received ECMO upon transfer4. The larger and more recent EOLIA trial failed to demonstrate this improvement in mortality5. The conclusion I take from this is that treatment at a high volume center matters and that a boarding patient with refractory hypoxemia warrants an early consideration for transfer to a tertiary center if high-quality ARDS care can’t be accomplished upstairs at your shop.
References
Mohr NM, Wessman BT, Bassin B, et al. Boarding of Critically Ill Patients in the Emergency Department. Crit Care Med. 2020;48(8):1180-1187. doi:10.1097/CCM.0000000000004385
Sottile PD, Albers D, Smith BJ, Moss MM. Ventilator dyssynchrony – Detection, pathophysiology, and clinical relevance: A Narrative review. Ann Thorac Med. 2020;15(4):190. doi:10.4103/atm.ATM_63_20
McGurk K, Riveros T, Johnson N, Dyer S. A primer on proning in the emergency department. J Am Coll Emerg Physicians Open. 2020;1(6):1703-1708. doi:10.1002/emp2.12175
Peek GJ, Mugford M, Tiruvoipati R, et al. Efficacy and economic assessment of conventional ventilatory support versus extracorporeal membrane oxygenation for severe adult respiratory failure (CESAR): a multicentre randomised controlled trial. Lancet Lond Engl. 2009;374(9698):1351-1363. doi:10.1016/S0140-6736(09)61069-2
Combes A, Hajage D, Capellier G, et al. Extracorporeal Membrane Oxygenation for Severe Acute Respiratory Distress Syndrome. N Engl J Med. Published online May 23, 2018. doi:10.1056/NEJMoa1800385

Matt McCauley, MD
How To Cite This Post:
[Peer-Reviewed, Web Publication] King, P. Akehtuamhen, A. (2022, Feb 28). Crashing Ventilator Patient. [NUEM Blog. Expert Commentary by McCauley, M]. Retrieved from http://www.nuemblog.com/blog/crashing -vent-patient.
Other Posts You May Enjoy



Review of the ATHOS 3 trial

Written by: Saabir Kaskar, MD (NUEM ‘23) Edited by: Amanda Randolph, MD (NUEM ‘20)
Expert Commentary by: Matt McCauley, MD (NUEM’ 21)
Review of the ATHOS 3 Trial: Angiotensin II for the Treatment of Vasodilatory Shock
Angiotensin, first isolated in the late 1930s, in recent years has become the new innovative vasopressor used in intensive care units, a change driven largely by the results of the ATHOS-3 trial. The ATHOS-3 trial in 2017 explored the efficacy of angiotensin II as a vasopressor for severe vasodilatory shock. Severe shock is defined as persistent hypotension requiring vasopressors to maintain a mean arterial pressure of 65mmHg and serum lactate <2 despite adequate volume resuscitation. Two classes of vasopressors have been used in the past for hypotension. They are catecholamines and vasopressin-like peptides. The human body, however, employs a third class which is angiotensin. Angiotensin II is an octapeptide hormone and a potent vasopressor that is an integral component of the renin-angiotensin-aldosterone system. It works by activating the ANGII type 1 receptor which subsequently activates a G coupled protein pathway and phospholipase C, thereby inducing vasoconstriction.
The ATHOS-3 trial compared the efficacy and safety of angiotensin II versus placebo in catecholamine-resistant hypotension, which is defined as an inadequate response to standard doses of vasopressors. The study was designed as a phase III multicenter randomized placebo control trial taking place across 75 intensive care units in the United States from 2015 to 2017. The three main inclusion criteria were catecholamine-resistant hypotension (defined as >0.2ug/kg/min of norepinephrine or equivalent for 6-48 hours to maintain a MAP 55-70 mmHg), adequate volume resuscitation (25mL/kg of crystalloid), and features of vasodilatory shock (mixed venous O2 >70% and CVP >8mmHg or cardiac index >2.3 L/min/m2).
Patients in vasodilatory shock that met the criteria of catecholamine-resistant hypotension were randomized to treatment with angiotensin II or placebo. Angiotensin II was initiated at an infusion rate of 20ng/kg/min and adjusted during the first three hours to increase MAP to at least 75mmHg. The primary outcome of the study was the response in MAP three hours after the start of angiotensin II infusion. A response was deemed as a MAP increase of 10mmHg from baseline or a MAP over 75mmHg without an increase in baseline vasopressor infusions. During the first three hours, the angiotensin II group had a significantly greater increase in MAP than placebo (12.5mmHg vs 2.9 mmHg). Angiotensin II also allowed for rapid increases in MAP which permitted decreases in doses of baseline catecholamine vasopressor. Additionally, improvement in the cardiovascular SOFA score was significantly greater in the angiotensin II group than in the placebo group. However, the overall SOFA score did not differ between groups. Rates of adverse events such as tachyarrhythmias, distal ischemia, ventricular tachycardia, and atrial fibrillation were similar in the angiotensin II and placebo groups. Overall serious adverse events that included infectious, cardiac, respiratory, gastrointestinal, or neurologic events were reported in 60.7% of patients who received angiotensin II and 67.1% of patients who received placebo.
The strengths and limitations of the ATHOS 3 trial are critical to how its author’s conclusions should be interpreted. The strengths of the study include that it was a randomized double-blind control trial examining a new class of vasopressor for refractory vasodilatory shock. Refractory shock is a common condition with high mortality, and so the investigation of an additional treatment modality can be of great clinical impact. However, one limitation of the study was that it was underpowered to demonstrate a mortality difference. It showed improvement in blood pressure which is a clinically important parameter but not a patient-oriented outcome. Interestingly, when vasopressin was studied in 2008, it similarly did not show a mortality benefit when added to norepinephrine infusion in septic shock2. It did, however, show a decrease in norepinephrine dosing which parallels the findings of the ATHOS 3 trial.
An additional point of contention with the ATHOS 3 trial is that the manuscript does not report an increase in thrombotic risk. It has been shown that angiotensin II increases thrombin formation and impairs thrombolysis3. The FDA even reports angiotensin II has a risk for thrombosis as there was a higher incidence (13% vs 5%) of arterial and venous thrombotic events in the angiotensin II vs placebo group in the ATHOS 3 trial itself. For this reason, the FDA recommends concurrent VTE prophylaxis with the use of angiotensin II. Further data regarding the thrombotic risk of angiotensin II would be helpful to determine which patient populations the vasopressor should be avoided in.
Overall, the author’s conclusion in the ATHOS 3 trial is that angiotensin II increased blood pressure in patients with a vasodilatory shock that did not respond to high doses of conventional vasopressors. It has been shown to raise mean arterial pressure over 75 mm Hg or by an increase of 10 mm Hg within three hours. The ATHOS 3 trial, however, did not demonstrate a mortality benefit when using angiotensin II. Further studies are needed to elucidate whether Angiotensin II truly improves patient outcomes in vasodilatory shock.
Expert Commentary
Thank you for this great summary of the ATHOS 3 trial. While this trial paved the way for the clinical use of angiotensin II as a vasopressor, you’ve raised some salient points as to why we should approach this emerging intervention with skepticism. The biggest shortcoming in my mind is the primary outcome of the study; it’s not particularly impressive that a vasopressor resulted in higher blood pressures compared to a placebo. Mortality benefit is an extremely elusive goal in critical care research1 but that doesn’t discount the fact that ATHOS 3 wasn’t designed to demonstrate an improvement in any patient-oriented outcome. ICU length of stay, hospital length of stay, ventilator-dependent days, or rate of renal replacement therapy: these are all things that matter to our patients and to our health systems and they are more fruitful targets when we investigate interventions.
There’s been some study of angiotensin II in the years since it has landed in our hospital formularies and there has not been robust data supporting its use. Some of the most recent data come from a multi-center retrospective study that includes patients from Northwestern. This review of 270 patients receiving angiotensin II demonstrated that 67% of patients were able to maintain a MAP of 65 with stable or reduced vasopressor doses. Univariate analysis showed that these patients that responded did have a statistically significant mortality benefit over the patients deemed nonresponders (41% vs 25%)2. If we are going to find a benefit of this drug, further study predicting which patients will be responders is necessary but this study did note that patients already receiving vasopressin and those with lower lactates (6.5 vs 9.5) were more likely to respond. Outside of septic shock, there is interest in the use of angiotensin II in refractory vasoplegia associated with post-cardiac surgery3 and anti-hypertensive overdose4. These are, of course, only hypothesis-generating.
But what does that mean to us clinically in the ED and ICU? This data shows us that angiotensin II can make the blood pressure better but I would never let it distract you from the things we know matter in sepsis resuscitation. Source control timely antibiotics, rational fluid resuscitation, and ruling out other causes of vasopressor refractory shock to include anaphylaxis, hemorrhage, adrenal insufficiency, LVOT obstruction, and any other cause of cardiogenic shock need to be ruled out and addressed. In my personal practice, I make sure to optimize these and start vasopressin shortly after the initiation of norepinephrine. In a patient already on vaso that has stopped responding to escalating doses of norepinephrine, I reach for my ultrasound probe and reassure myself that there isn’t significant sepsis-related myocardial dysfunction because those patients may benefit from a trial of an inotrope like epinephrine. In those with a good cardiac squeeze, I think it’s appropriate to discuss with your intensivist and clinical pharmacist the utility of adding angiotensin II as part of a kitchen-sink approach. Until we have more data about the benefits of this extremely expensive intervention, I wouldn’t lose sleep if you’re unable to secure it for your patient.
References
Chawla LS et al. Intravenous Angiotensin II for the Treatment of High-Output Shock (ATHOS Trial): A Pilot Study. Crit Care 2014; 18(5): 534. PMID: 25286986
Russell JA et al. Vasopressin Versus Norepinephrine Infusion in Patients with Septic Shock. NEJM 2008; 358(9): 877 – 87. PMID: 18305265
Celi A et al. Angiotensin II, Tissue Factor and the Thrombotic Paradox of Hypertension. Expert Review of Cardiovascular Therapy 2010; 8(12): 1723-9 PMID: 21108554
Santacruz CA, Pereira AJ, Celis E, Vincent JL. Which Multicenter Randomized Controlled Trials in Critical Care Medicine Have Shown Reduced Mortality? A Systematic Review. Crit Care Med. 2019;47(12):1680-1691. doi:10.1097/CCM.0000000000004000
Wieruszewski PM, Wittwer ED, Kashani KB, et al. Angiotensin II Infusion for Shock: A Multicenter Study of Postmarketing Use. Chest. 2021;159(2):596-605. doi:10.1016/j.chest.2020.08.2074
Papazisi O, Palmen M, Danser AHJ. The Use of Angiotensin II for the Treatment of Post-cardiopulmonary Bypass Vasoplegia. Cardiovasc Drugs Ther. Published online October 21, 2020. doi:10.1007/s10557-020-07098-3
Carpenter JE, Murray BP, Saghafi R, et al. Successful Treatment of Antihypertensive Overdose Using Intravenous Angiotensin II. J Emerg Med. 2019;57(3):339-344. doi:10.1016/j.jemermed.2019.05.027

Matt McCauley, MD
How To Cite This Post:
[Peer-Reviewed, Web Publication] Kaskar, S. Randolph, A. (2022, Feb 14). Review of ATHOS 3 trial. [NUEM Blog. Expert Commentary by McCauley, M]. Retrieved from http://www.nuemblog.com/blog/review-athos3-trial.
Other Posts You May Enjoy


Vasopressor Nonresponse

Written by: Elizabeth Stulpin, MD (NUEM ‘23) Edited by: Aaron Wibberly, MD (NUEM ‘22)
Expert Commentary by: Joshua Zimmerman, MD (NUEM ‘17)
Non-Response to Vasopressors
Shock is defined as a state of cellular and tissue hypoxia resulting in end organ dysfunction. This state may arise due to impaired oxygen delivery to tissues, impaired oxygen utilization by the tissues themselves, increased oxygen consumption, or a combination of these mechanisms. Due to its extremely high morbidity and mortality as well as high healthcare costs, the prompt recognition, diagnosis and resuscitation of shock is key. And for most forms, EM physicians are not typically shocked by shock. They have a toolbox of strategies, mainly fluids and vasopressors, to stabilize these critically ill patients.
However, what happens when the trusted treatment paradigm fails? There is a subset of patients who, despite aggressive conventional resuscitation, have an inadequate hemodynamic response and develop refractory shock. This is seen in approximately 7 percent of patients, with short-term mortality ranging from 50 to 80 percent. Due to this significantly lower incidence and increased mortality, alternate causes for refractory shock must be considered when vasopressors do not have the desired effect.
Acidosis
Acidosis in shock states can present from multiple different sources, including sepsis, hypoxemia, ingestions, hyperlactatemia from hypoperfusion, amongst others. With increasing acidosis, calcium influx is reduced, contractility is inhibited and the binding affinity of pressors is reduced, all of which lead to excess vasodilation and refractory hypotension. While bicarbonate is sometimes given in an effort to increase cellular pH, it is controversial for any pH >7.0. At those levels, bicarbonate administration has not been shown to improve cardiac output, MAP or pressor response. While a bicarbonate drip and hyperventilation can temporize an acidosis, emergent HD or CRRT is a definitive treatment if the cause cannot be quickly reversed.
Adrenal Insufficiency
Cortisol has a myriad of functions in the body, not limited to its synergistic effects with catecholamines to help cause vasoconstriction. Thus, when the adrenal glands are chronically suppressed and then experience an acute stressor, hypotension can ensue. The most common cause of chronic suppression is long-term steroid use, and a stressor can include surgery, infection, hypovolemia, pregnancy, medications, or reduced steroid use. Clues that suggest adrenal insufficiency include nausea/vomiting, cutaneous hyperpigmentation, and multiple electrolyte abnormalities (hyponatremia, hyperkalemia, hypoglycemia). Previously healthy individuals, in the setting of critical illness, can also infrequently decompensate into a state of relative adrenal insufficiency. To reverse these effects as well as refractory hypotension, hydrocortisone is the preferred agent due to both its glucocorticoid and mineralocorticoid properties. A loading dose of 100mg IV should be given, followed by 50mg every 6 hours thereafter.
Alternate Shock
Not all shock is declared equal. For example, a patient in cardiogenic shock will likely worsen with the administration of fluids and the wrong vasopressors. Similarly, obstructive shock as seen in massive PEs, tension pneumothoraces or cardiac tamponade will not improve without addressing the cause.
Anaphylaxis
Anaphylaxis may present as hypotension alone. Thus, it may easily be confused with a different form of shock and treated with vasopressors such as norepinephrine and vasopressin, which are not first line for anaphylaxis. Along with using epinephrine as the pressor of choice and other conventional therapies for anaphylaxis, there are alternate medications available for persistent refractory hypotension. One of these is methylene blue. While typically reserved for treatment of methemoglobinemia, the cellular mechanism of methylene blue can decrease vasodilation. Data suggest that this effect can be seen with a one-time dose of 1-2mg/kg.
Hemorrhage
Often a common cause of refractory shock in the post op setting, bleeding can be obscure in its early stages before a hemoglobin drop is appreciated or before the patient develops abdominal distension and flank dullness (retroperitoneal bleed). If concerned, empiric uncrossed unmatched blood can be transfused.
Hypocalcemia
Calcium homeostasis is necessary for the proper maintenance of myocardial contractility and vascular tone. Hypocalcemia can be hinted at through history or by hints such as a prolonged QTc on an ECG. Those at higher risk of hypocalcemia (vitamin D deficiency, ESRD, hyperparathyroidism, burns, multiple blood transfusions, etc.) may have greater severity of shock with increased mortality. In repleting calcium, co-administration of phosphorous and magnesium may allow for reversal of the patient’s shock state.
Hypothyroidism
Decompensated hypothyroidism can have profound effects, including bradycardia, impaired myocardial contractility, and decreased peripheral vasoconstriction. However, even subclinical hypothyroidism with an elevated TSH but normal T4 increases risk of poor outcomes due to effects on cardiac function. While the diagnosis of hypothyroidism can be delayed by lab results, clues to the diagnosis include thyroidectomy scar, non-pitting edema of the extremities, macroglossia, altered mental status, hypoglycemia and hypothermia. Initial hypotension may not respond to vasopressors, but shock should improve once thyroid hormone is given. Stress dose steroids (hydrocortisone 100mg IV) should also be given due to the association between adrenal insufficiency and hypothyroidism, and giving thyroid hormone without steroids can precipitate adrenal crisis.
Ingestions
When in doubt, look at the medication list! Both beta blocker and calcium channel blocker toxicity can cause profound myocardial depression, bradycardia and hypotension due to their inhibition of calcium signaling. Refractory hypotension can be overcome with the use of direct cardiac pacing, calcium, glucagon, or high dose insulin. And if all else fails, ECMO can overcome medication toxicity until it can be fully metabolized or cleared.
When conventional resuscitation for shock with fluids, vasopressors and/or inotropes fails, it is time for a cognitive pause. By running through this list of alternate causes of refractory shock, other methods of resuscitation can be added to improve patient outcomes and stabilize the patient.
Sources:
1) Amrein K, Martucci G, and Hahner S. Understanding adrenal crisis. Intensive Care Medicine. 2018; 44(5): 652-655.
2) Boyd J, Walley K. Is there a role for sodium bicarbonate in treating lactic acidosis from shock? Curr Opin Crit Care. 2008;14:379-83.
3) Farkas J. Decompensated Hypothyroidism. The Internet Book of Critical Care. 2016. Accessed https://emcrit.org/ibcc/myxedema/
4) Ho H, Chapital A, and Yu M. Hypothyroidism and Adrenal Insufficiency in Sepsis and Hemorrhagic Shock. Arch Surgery. 2004; 139(11):1199-1203.
5) Kerns W. Management of B-Adrenergic Blocker and Calcium Channel Antagonist Toxicity. Emergency Medicine Clinics of North America. 2007; 25: 309-331.
6) Levy B, Collin S, Sennoun N, et. al. Vascular hyporesponsiveness to vasopressors in septic shock: from bench to bedside. Intensive Care Medicine. 2010; 36: 2019-2029.
7) Manji F, Wierstra B, and Posadas J. Severe Undifferentiated Vasoplegic Shock Refractory to Vasoactive Agents Treated with Methylene Blue. Case Reports in Critical Care. 2017.
8) Minisola, S et al. Serum Calcium Values and Refractory Vasodilatory Shock. Chest. 2019; 155(1): 242.
9) Nandhabalan P, Ioannou N, Meadows C and Wyncoll D. Refractory septic shock: our pragmatic approach. Critical Care. 2018; 22(1):215.
10) Smith L, and Branson B. Refractory Hypotension – Diagnosis and Management in Surgical Patients. California Medicine. 1961; 95(3): 150-155
11) Velissaris D, Karamouzos V, Ktemopoulos N, Pierrakos C, and Karanikolas M. The Use of Sodium Bicarbonate in the Treatment of Acidosis in Sepsis: A Literature Update on a Long Term Debate. Critical Care Research and Practice. 2015.
12) Wang H, Jones A, and Donnelly J. Revised National Estimates of Emergency Department Visits for Sepsis in the United States. Critical Care Medicine. 2017; 45(9): 1443-1449.
Expert Commentary
Dr. Stulpin's review of a very critical topic is well articulated and concise. I would like to particularly emphasize her final points before delving into the details. As she pointed out, patients failing to respond to typical resuscitative efforts represent quite a quagmire to the ED physician. It is easy to arrive at premature closure and presume all shock to be sepsis simply needing more fluids or vasopressors. However, there is significant risk in this practice, and I would thus advocate very strongly for a “pause” and thorough reassessment of the patient’s presentation and condition. Much akin to a pre-procedural time out, this should be deliberate and uninterrupted to prevent diagnostic momentum from building and arriving at a premature closure on the etiology of the patient’s condition.
This review covers much of the differential and pathophysiology that is germane to a discussion of refractory shock. Rather than review this in detail, I would like to discuss a practical approach to application of this knowledge at the bedside. So, as we approach the patient who is refractory to standard resuscitation, one’s first task is to confirm the patient is receiving the desire therapy and that this is truly shock refractory to intervention. Check all access points for infiltration –the single most reversible cause for refractory hypotension is that the patient is not receiving fluids/vasopressors, etc. due to inadequate access. Check dosing and confirm with nursing that infusions have been running appropriately. If access is the issue, I would advocate placing a more reliable form such as central access or, in the very unstable, IO access early in the reassessment. While I am an advocate for US guided access in stable patients and feel that this is often a great tool, this is a scenario I would advocate against US guided peripheral access. This form of access is often time consuming, and more importantly, signs of infiltration are less evident in deeper IV sites which is a serious concern if you are running peripheral vasopressors.
Presuming your access is adequate, the next task is to reassess perfusion in its entirety. While not mandatory, this is a point where I would consider placing an arterial line for optimally reliable BP monitoring. One must perform a complete re-examination as well. If not already completed, a rectal exam for the occult GI bleeding. One should additionally particular attention on reassessment to skin and perfusion. Check capillary refill, skin temperature and coloration as a matter of habit for cases of refractory shock. Cool extremities are not typically associated with distributive shock and should make you consider shock states with high SVR such as cardiogenic or acute blood loss. In addition, a repeat skin exam may reveal new rashes – I look specifically for the presence of petechiae, purpura or urticaria. Urticaria should prompt consideration of anaphylaxis. If the patient has received any antibiotics yet this is an important consideration given antibiotics are one of the most common medication precipitants of anaphylaxis and this may be the source of their worsening shock state.
After completing your thorough exam, I strongly advocate for a sonographic assessment as well. This is where the RUSH exam, or at least a modified version, fits well into patient assessment and can offer a great deal of information. I pay particularly close attention to the cardiac and abdominal windows. This can aid in the rapid diagnosis of an obstructive shock state such as cardiac tamponade, or acute RV failure in the setting of massive PE. Free intra-abdominal fluid should be considered hemorrhage until proven otherwise in the shock patient. One reminder is that when assessing for occult bleeding, the FAST exam views are an excellent tool but not sufficiently sensitive for definitive rule out. In particular, the FAST exam lacks sensitivity for retroperitoneal bleeding and therefore if there remains high clinical concern, CT imaging should also be considered.
Once a thorough re-examination is completed, reconsider the patient’s medications as another source. Patients are on a myriad of antihypertensives and other agents that can lower blood pressure. Many of the pathways that metabolize or eliminate these drugs are compromised in a state of hypoperfusion and can lead to a synergistic worsening of hypotension. One example of this is the case of BRASH Syndrome. In addition to antihypertensives, a review of the patient’s medication list should include a particular search for steroids or other adrenal replacements such as fludrocortisone. An extremely important cause of refractory hypotension to consider is that of adrenal insufficiency. As Dr. Stulpin reviewed in her discussion above, this can come in both primary and secondary forms. The latter is far more common and induced by exogenous steroid use. A wise ICU attending once taught that no patient should die without stress dose steroids. While perhaps a bit morbid, the take home point here was that adrenal insufficiency can present in any patient, not just those with underlying disorders of glucocorticoid production. Pay particular attention to patients with autoimmune disorders or others on chronic steroid therapy as these are the populations that are particularly at risk. Patients presenting with unexplained hypoglycemia in the setting of sepsis or shock of any kind should also be strongly considered to receive stress dose steroids. Do not wait for a cortisol level to treat this condition. Consider, as well, checking TSH and free T3/T4 as patients with adrenal insufficiency can simultaneously harbor other endocrinopathies.
To summarize, refractory hypotension or failure to respond to traditional interventions is relatively uncommon but critical to identify in shock patients. Often these patients have a primary diagnosis of septic shock but can suffer from concurrent shock related to one or more of the differential considerations we have reviewed above. A thoughtful reassessment, both of the patient’s physical exam findings, US and other diagnostics, medication list and history of present illness will offer clues that may uncover an additional etiology critical to treat and to ensure the best outcome possible.

Joshua D. Zimmerman, MD
Health System Clinician
Feinberg School of Medicine
Northwestern Medicine
How To Cite This Post:
[Peer-Reviewed, Web Publication] Stulpin, E. Wibberly, A. (2022, Jan 24). Vasopressor Nonresponse. [NUEM Blog. Expert Commentary by Zimmerman, J]. Retrieved from http://www.nuemblog.com/blog/vasopressor-nonresponse
Other Posts You May Enjoy



Bicarb in Cardiac Arrest

Written by: Kishan Ughreja, MD (NUEM ‘23) Edited by: Sean Watts, MD (NUEM ‘22)
Expert Commentary by: Dana Loke, MD (NUEM ‘21)
Utility of Sodium Bicarbonate in Cardiac Arrest
Use of sodium bicarbonate as empiric therapy in cardiac arrest has been an area of controversy. During cardiac arrest hypoxia and hypoperfusion results in severe metabolic acidosis and subsequent impaired myocardial contractility, decreased efficacy of vasopressors, and increased risk of dysrhythmias. Previous ACLS guidelines recommended use of sodium bicarbonate to mitigate these effects; however, harms are also associated with its routine use including compensatory respiratory acidosis, hyperosmolarity, increased vascular resistance, and reduction in ionized calcium. 1 Current guidelines no longer recommend routine use of sodium bicarbonate, except in cases of arrest secondary to hyperkalemia, TCA overdose or preexisting metabolic acidosis.2 Regardless of these recommendations, sodium bicarbonate continues to be utilized during routine management of cardiac arrest, and studies are limited in investigating its appropriate use.
The study below investigates the effect of sodium bicarbonate in patients suffering out-of-hospital cardiac arrest with severe metabolic acidosis during prolonged CPR.
Article

Clinical Question
In patients with prolonged, atraumatic out-of-hospital cardiac arrest (OHCA) and severe metabolic acidosis, does sodium bicarbonate (SB) administration with transient hyperventilation improve acidosis without increased CO2 burden, enhance rates of return of spontaneous circulation (ROSC), survival to admission, and favorable neurologic outcomes?
Study Design
Double-blind, prospective, randomized, placebo-controlled, single-center pilot clinical trial
Population
Inclusion criteria: Atraumatic arrest in patients ≥18yo without ROSC after 10 minutes of CPR in ED and with pH <7.1 or bicarbonate <10 mEq/L on ABG
Exclusion criteria: DNR, ECPR, ROSC w/i 10 minutes of ACLS, absence of severe metabolic acidosis on ABG after 10 minutes of CPR
Data collection over 1 year at Asan Medical Center, a tertiary referral center in Seoul, Korea
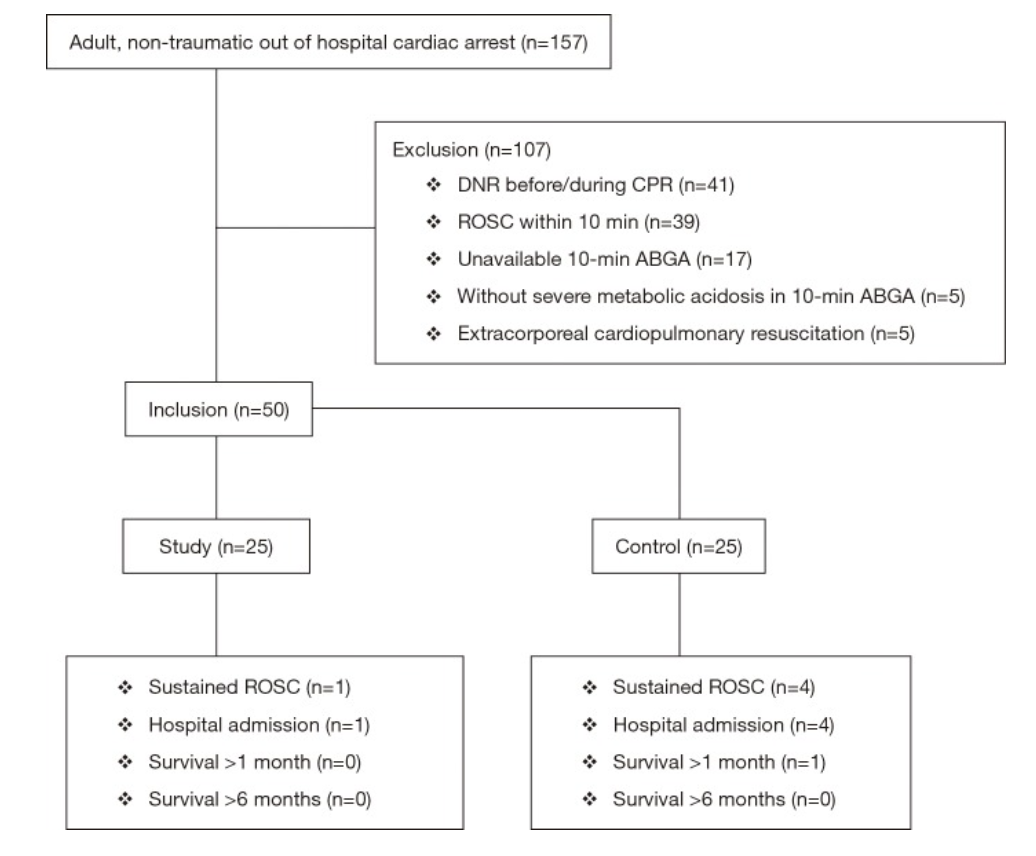
Figure 1: Patient Selection
Intervention
Sodium bicarbonate administration of 50 mEq/L over 2 minutes with concurrent increase in ventilation rate from 10 to 20 breaths per minute for 2 minutes
Control
Normal saline administration of 50 mL over 2 minutes (with same transient hyperventilation)
Outcomes
Primary
Change in acidosis (per methods section)
Secondary
Sustained ROSC — defined as restoration of a palpable pulse ≥20 min (per methods section, but listed as primary outcome in abstract)
Survival to hospital admission
Good neurological survival at 1 and 6 months (defined as cerebral performance category 1 or 2)
Results
157 patients presented with cardiac arrest, 50 enrolled per inclusion criteria
No significant differences between study and control groups regarding demographics, PMH, witnessed arrest, bystander CPR, pre-hospital and initial cardiac rhythm
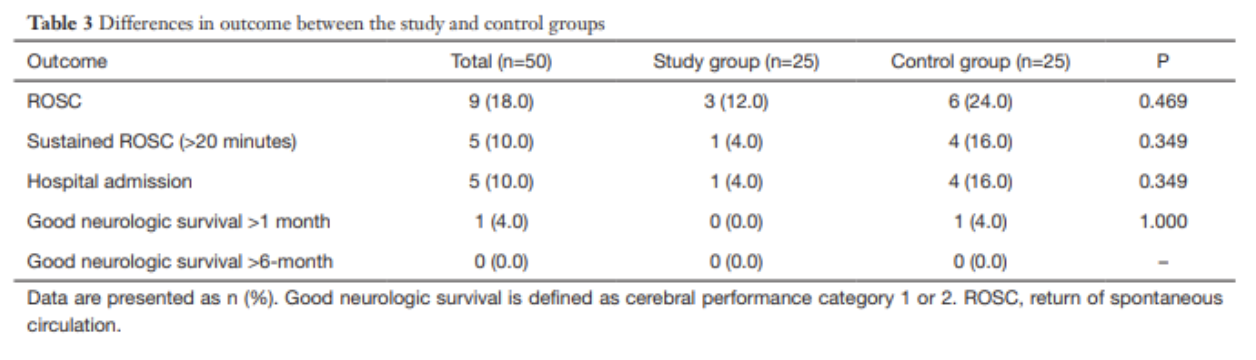
10% (n=5) of enrolled patients with sustained ROSC and admitted
No patients survived at 6 months follow up

Pre-Intervention
ABG results at 10 minutes were not significantly different between groups
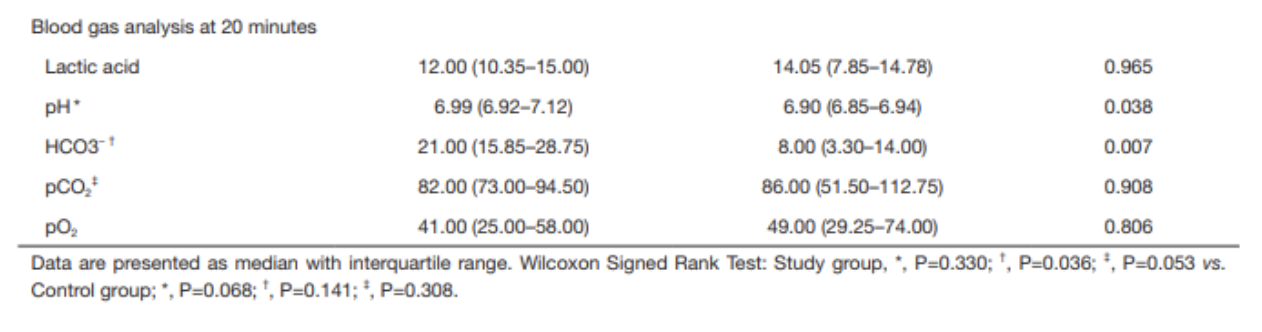
Post-intervention
ABG results at 20 minutes demonstrate that pH and HCO3- were higher in the study group than in the control group
pH 6.99 vs 6.90, p=0.038
HCO3- 21.0 vs 8.00, p=0.007
Within the study group, the increase in pH was not statistically significant after sodium bicarbonate administration; the increase in HCO3- was statistically significant (using Wilcoxon signed rank test)
No statistically significant findings in the control group after normal saline administration
No significant differences in any secondary outcomes (sustained ROSC, survival to admission, good neurologic outcome)
Strengths
Randomized, double-blinded, placebo-controlled study design
This study adds additional information to a clinical question that has limited previous research
This study added a practical clinical intervention (hyperventilation) to counteract excessive CO2 accumulation secondary to sodium bicarbonate administration, a known deleterious effect of this compound.
Strong control over sodium bicarbonate administration (no pre-hospital administration allowed in South Korea), so authors could control when it was given and analyze ABG results at desired intervals)
Weaknesses
Small, single-center study with only 50 enrolled patients
Primary endpoint unclear from abstract vs methods, whether it was change acidosis or sustained ROSC; however, neither is truly patient-centered clinical outcome (good neurological outcome would be the ideal primary outcome)
Dosing was universal — 50 mEq/L instead of weight based (1-2 mEq/L/kg), which could result in improper dosing
Hyperventilation strategy may have benefited sodium bicarbonate administration group by countering respiratory alkalosis, however, it could have harmed the placebo group
Possible venous sampling rather than arterial for blood gas analysis at 10-minute point, though this would be a concern in any arrest setting if an arterial line could not be established in this time frame
Author’s Conclusion
“The use of sodium bicarbonate during CPR with transient hyperventilation improves acid-base status without CO2 elevation which is one of the most concerned adverse effects of sodium bicarbonate administration, but it had no effect on the improvement of the rate of ROSC and good neurologic survival. At this point, we could not advise for or against its administration, our pilot data could be used to help design a larger trial to verify the efficacy of sodium bicarbonate.”
Bottom Line
Based on this study, the use of sodium bicarbonate does not appear to improve clinically significant outcomes, though it improved acid-base status. Sodium bicarbonate should not be indiscriminately used in all cardiac arrests, and larger trials should be performed to further evaluate its impact on patient-centered outcomes.
Citation
Ahn, S., Kim, Y. J., Sohn, C. H., Seo, D. W., Lim, K. S., Donnino, M. W., & Kim, W. Y. (2018). Sodium bicarbonate on severe metabolic acidosis during prolonged cardiopulmonary resuscitation: a double-blind, randomized, placebo-controlled pilot study. Journal of thoracic disease, 10(4), 2295.
References
White, S. J., Himes, D., Rouhani, M., & Slovis, C. M. (2001). Selected controversies in cardiopulmonary resuscitation. Seminars in respiratory and critical care medicine, 22(1), 35–50. https://doi.org/10.1055/s-2001-13839
Merchant, R. M., Topjian, A. A., Panchal, A. R., Cheng, A., Aziz, K., Berg, K. M., Lavonas, E. J., Magid, D. J., & Adult Basic and Advanced Life Support, Pediatric Basic and Advanced Life Support, Neonatal Life Support, Resuscitation Education Science, and Systems of Care Writing Groups (2020). Part 1: Executive Summary: 2020 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation, 142(16_suppl_2), S337–S357. https://doi.org/10.1161/CIR.0000000000000918
Expert Commentary
Thank you Dr. Ughreja and Dr. Watts for this excellent blog post on an important topic. In medicine, we often ask “what else can we do?” but less often do we ask “is what we’re already doing effective?” This is especially important for resuscitation and cardiac arrest. Not everything that is standard-of-care is ultimately effective care, and overtreating patients can lead to other untoward effects.
In addition to the points made in the above blog, I would add a few important notes into the equation. First, the study excluded in-hospital cardiac arrest and therefore should not be considered in those patients. Second, the study also excluded those patients with early ROSC and absence of severe metabolic acidosis, effectively biasing towards inclusion of sicker patients. It is unclear how administration of sodium bicarbonate may have influenced those patients. Third, the study population was quite small and a striking majority of that population were found to have an initial rhythm of asystole. Fourth, ventilation rates were purposefully increased during bicarb administration. Though this may be practical and can potentially counteract excessive CO2 accumulation secondary to sodium bicarbonate administration, this is not common practice which leads to questions of this study’s external validity at other institutions.
So, despite this study, at this point in time we still must grapple with the “should-we-or-should-we-not” of sodium bicarbonate administration in prolonged cardiac arrest. Some scenarios certainly do require sodium bicarbonate, most notably TCA overdose and hyperkalemia. In these cases, it’s obvious what to do. But so often what we do in emergency medicine is riddled with uncertainty. An unclear cause of cardiac arrest is certainly one of those situations. Perhaps instead of mindlessly giving sodium bicarbonate to cardiac arrest patients, we should give it once or twice and look for evidence that it has had an effect. Is the rhythm narrowing? Did you obtain ROSC shortly after administration? If not, giving dose after dose of sodium bicarbonate in hopes of meaningful recovery may not be the best path forward.

Dana Loke, MD
Department of Emergency Medicine
Northwestern University Feinberg School of Medicine
Northwestern Memorial Hospital
How To Cite This Post:
[Peer-Reviewed, Web Publication] Ughreja, K. Watts, S. (2021, Dec 6). Bicarb in Cardiac Arrest. [NUEM Blog. Expert Commentary by Loke, D]. Retrieved from http://www.nuemblog.com/blog/bicarb-arrest
Other Posts You May Enjoy

Mechanical Ventilation Oversimplified
Written by: Shawn Luo, MD (NUEM ‘22) Edited by: Sam Stark, MD, MA (NUEM ‘20)
Expert Commentary by: Ruben Mylvaganam, MD
The ventilator: we’ve all learned about it - the lectures, the bed-side demonstrations on those mind-numbingly long ICU rounds. But we were also told, repeatedly, “Don’t touch it!” Unless you are an attending, fellow, or respiratory therapist (RT) of course. So for a lot of us, the ventilator is a black box, mythical and intimidating.
In this blog, I hope to demystify ventilators a little so when duty calls, you can set initial settings and make some basic adjustments.
Physiology
1. How Mechanical Ventilation affects Oxygenation: PEEP & FiO2
You can reference this nice ARDSnet table for FiO2/PEEP combinations.

FiO2 – its effect is immediate
PEEP – takes up to an hour to show full effect
Therefore, when weaning, wean FiO2 before weaning PEEP so that if the patient desaturates, you have room to go up on FiO2.
2. How Mechanical Ventilation affects Ventilation: Tidal Volume, Respiratory Rate, Inspiratory Pressure or Inspiratory Time
This should be titrated in response to the patient's CO2 levels. Patients in respiratory failure from profound metabolic acidosis will need you to set higher minute ventilation to attempt respiratory compensation.
3. Peak Pressure and Plateau Pressure
Peak pressure is the summation of both airway resistance (dynamic compliance) and plateau pressure (static compliance). Most modern ventilators will automatically report peak pressures without any special maneuvers required. When thinking about airway resistance, think of when you blow air through a straw – the narrower the tubing the higher the resistance and thus a lot of pressure is needed to generate that flow. To measure airway resistance, have the RT set the flow rate to 60 LPM, adjust the flow pattern to a square wave form, and ask them to perform an inspiratory hold.
Plateau pressure is related to lung compliance (higher plateau pressure = less compliant lung). It is the pressure “felt” by the alveoli, and keeping it less than 30 cm H2O helps to prevent barotrauma. It’s only measured after the air stops moving (via an inspiratory hold maneuver – ask RT how to do this on your ventilator) so that dynamic airway resistance is not a factor.
4. Breath-stacking / Auto-PEEP
This occurs when the patient does not have enough time to finish exhalation before the next breath is delivered. This results in progressive hyperinflation of the lung, high peak pressures, and eventually hemodynamic collapse if not identified and intervened upon. It is most common in obstructive airway diseases such as asthma and COPD. Be vigilant for the flow diagram below on the ventilator to detect it early.

Modes
Volume vs Pressure – WHAT TYPE of breath is targeted
Volume mode means the vent will deliver a set tidal volume of air and results in whatever pressure (i.e. stiffer lungs result in higher pressure)
Pressure mode in turn means the vent will deliver at a set inspiratory pressure, and results in whatever volume (i.e. stiffer lungs result in lower volume)
A/C (Assist/Control) vs Support – WHEN the breath is delivered
In A/C mode, the machine delivers breath at a pre-set frequency (control), but the patient can also trigger additional breaths (assist) to faster than the set frequency. A quick and dirty trick is that any mode that contains the word “Control” means there will be a minimal respiratory rate set by the clinician.
Support (or Spontaneous) mode, in turn, will only deliver a breath when the patient initiates it. It senses the negative pressure generated by the patient and delivers a breath. If the patient does not breathe, it will not deliver. Usually safety back-up is in place to prevent prolonged apnea.

Volume Control
Delivers set tidal volume at or above a set rate
You set: tidal volume (6-8mL/kg ideal body weight), respiratory rate (16-22 breaths per minute), flow rate (60-80 LPM), and PEEP & FiO2 as needed
Check: Plateau pressure <30 (inspiratory hold maneuver)
This is a good initial setting for most of the patients you just intubated
Pressure Control
Delivers set pressure at or above set rate
You set: inspiratory pressure (5-15 cm H2O), inspiratory time (“I-time”; 0.6-0.8), respiratory rate (16-22), PEEP & FiO2 as needed
Check: to make sure the patient is getting tidal volumes of 6-8 mL/kg
This can be a helpful setting in some patients that do not tolerate volume control. Adjust pressure support to achieve tidal volume of 6-8 mL/kg while ensuring total pressure is less than 30-35 cm H20.
Pressure Support
Delivers set pressure when the patient initiates a breath to help the patient move the air
You set: Pressure support (5-15 cm H2O), PEEP & FiO2 as needed
Check: to make sure the patient is getting tidal volumes of 6-8 mL/kg
Usually a weaning mode to check if the patient is likely to tolerate extubation
*The bottom line is, by adjusting the parameters, you can achieve the same result with different ventilation modes.
My step-wise approach to initiate mechanical ventilation on most patients:
Build initial settings around Volume Control (tidal volume 6-8mL/kg ideal body weight, respiratory rate 16-22, PEEP 5, FiO2 100%)
Tweak according to patient’s clinical scenario – e.g. higher respiratory rate for acidotic patients, higher initial PEEP for hypoxemic respiratory failure, longer expiratory time for asthmatics/COPD patients with auto-PEEP
Start mechanical ventilation, quickly wean FiO2 for a goal SpO2 of 94-98%
Adjust settings further based on clinical response and ABGs
When in doubt, disconnect and bag the patient.
References:
The Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000;342:1301-1308.
Weingart, S. Managing Initial Mechanical Ventilation in the Emergency Department. Annals of Emergency Medicine, Volume 68, Issue 5, November 2016, Pg 614-617
Hyzy, R. Modes of Mechanical Ventilation. In: UpToDate, Parsons P. Finlay G (Ed), UpToDate, Waltham, MA. (Accessed on May 5, 2020.)
Expert Commentary
Thank you for the opportunity to review this very helpful and concise review on the basics of invasive mechanical ventilation. I hope to make this commentary brief, a contrast to our notoriously long ICU rounding habits. I would recommend any reader to view this editorial for a more in depth and nuanced understanding of mechanical ventilation. (1)
As you have described above, one way in which to think about mechanical ventilation is in the context of the most common scenarios in which we implement it, ie: hypoxemia and hypercapnia. Understanding that for hypoxemic patients, our tools to improve physiology are by manipulating the set FiO2 and PEEP to achieve specified targets for oxyhemoglobin saturation or P/F ratios (with regard to ARDS management). It is important to note that a few studies have demonstrated that an FiO2 greater than 50-60% can be toxic and may result in an increase in reactive oxygen species, increased airway damage (tracheobronchitis), and secondary infection from impaired bactericidal action of immune cells. (2,3) For our hypercapnic patients, knowing their prior baseline PCO2 is helpful in determining how to adjust the respiratory rate and tidal volume to appropriately improve their respiratory acidosis.
An important common 3 part methodology to better appreciate modes of mechanical ventilation is understanding the “trigger”, “target”, and “cycle” of each ventilator mode. In the simplest of terms, the “trigger” is what prompts the ventilator to deliver the breath (ie: an assisted breath when the ventilator senses a patient generated decrease in flow/pressure or a control breath when enough time has elapsed as mandated by the set respiratory rate). The “target” is what the ventilator aims to achieve with each breath (in the mode of AC-VC: a targeted flow rate [often ~60 L/min] or in the mode of AC-PC: a targeted inspiratory pressure [often ~15 cwp]). Finally, the “cycle” is a term that describes how the ventilator recognizes when it is time to terminate the breath that is delivered (in the mode of AC-VC: cycling off after the goal TV is reached [~600cc] or in the mode of AC-PC: cycling off after the set inspiratory time has occurred [~ 0.7 seconds]). See table below for a quick summary.
Finally, the best practical way to simplify mechanical ventilation is to request the changes by the respiratory therapist and see the effects. I encourage you to interpret all VBGs and ABGs, approach your respiratory therapist, pulmonary/CCM fellow, and suggest everything from initial ventilator settings, changes to both modes and individual parameter settings, and see the reflection of this work in your subsequent blood gases.

References
1. Walter JM, Corbridge TC, Singer BD. Invasive Mechanical Ventilation. South Med J. 2018 Dec;111(12):746-753. doi: 10.14423/SMJ.0000000000000905. PMID: 30512128; PMCID: PMC6284234.
2. Suttorp N, Simon LM. Decreased bactericidal function and impaired respiratory burst in lung macrophages after sustained in vitro hyperoxia. Am Rev Respir Dis. 1983 Sep;128(3):486-90. doi: 10.1164/arrd.1983.128.3.486. PMID: 6311064.
3. Griffith DE, Garcia JG, James HL, Callahan KS, Iriana S, Holiday D. Hyperoxic exposure in humans. Effects of 50 percent oxygen on alveolar macrophage leukotriene B4 synthesis. Chest. 1992 Feb;101(2):392-7. doi: 10.1378/chest.101.2.392. PMID: 1310457.

Ruben Mylvaganam, MD
Instructor of Medicine
Department of Pulmonology & Critical Care Medicine
Northwestern Memorial Hospital
How To Cite This Post:
[Peer-Reviewed, Web Publication] Luo, S. Stark, S. (2021, Nov 1). Mechanical Ventilation Oversimplified. [NUEM Blog. Expert Commentary by Mylvaganam, R]. Retrieved from http://www.nuemblog.com/blog/mechanical-ventilation-tips
Other Posts You May Enjoy
Hanging Injuries

Written by: Vytas Karalius, MD, MPH (NUEM ‘22) Edited by: Nery Porras, MD (NUEM ‘21) Expert Commentary by: Kevin Emmerich, MD, MS
Today’s post was inspired by the near-hanging of young gentleman who ended up passing away due to complications related to his near-hanging. His parents decided to donate his organs to Gift of Hope, allowing the passing of his life to extend the lives of others. While we hope to never see cases like these, they are an inevitable part of our job as emergency medicine physicians. As with most rare and complex pathology, preparation and knowledge can help us with the management of these cases when things often get chaotic. Lastly, as emergency medicine physicians who see the sequelae of mental illness daily in their EDs, I encourage us all to advocate for better funding and access to mental health care in the United States.
Hanging Injury
Terms/Classification [1]
“Hanging” is used to describe a death from a form of strangulation that involves hanging from the neck.
“Near-hanging” is a term for patients who have survived an attempted hanging (or at least long enough to reach the hospital).
“Complete hanging” defines when a patient’s legs are fully suspended off the ground and the patient's bodyweight is fully suspended by the neck.
“Incomplete hanging” defines when some part of the patient’s body is still on the ground and the body's full weight is not suspended off the ground.
“Judicial hanging” classically refers to victims who fell at least the height of their body.
Epidemiology:
Hanging is the 2nd most common form of successful suicide in the US after firearms
Accounts for 23% of >34,500 suicides in 2007
In the jail system, hanging is the most common form of successful suicide
Increasing incidence in US
Risk Factors: male, aged 15-44 years, history of drug or alcohol abuse, history of psychiatric illness
Pathophysiology of Injury:
Spine/Spinal Cord:
When the drop is greater than or equal to the height of the victim, as in a judicial hanging, there will almost always be cervical spine injury.
The head hyperextends, leading to fracture of the upper cervical spine ("hangman's fracture” of C2) and transection of the spinal cord.
Cervical injuries are in non-judicial hangings are rare. [2] One retrospective case review of near-hangings over a 10-year period found the incidence of cervical spine fracture to be as low as 5%. [3]
Vascular:
The major pathologic mechanism of death in hanging/strangulation is neck vessel occlusion, not airway obstruction. [1,4]
Death ultimately results from cerebral hypoxia and global ischemia.
There are two mechanisms by which this happens:
Venous: The most implicated cause of death is actually venous obstruction. Jugular veins are superficial and easily compressible. Obstruction of venous outflow from the brain leads to stagnant hypoxia and loss of consciousness in as little as 15 seconds.
Arterial: The risk of damage to the major arterial blood flow to the brain (such as carotid artery dissection) is rare, but should suspected in patients. [4]
Cardiac:
Carotid body reflex-mediated cardiac dysrhythmias are reported, and likely a minor mechanism of death.
Pulmonary:
Airway compromise plays less of a role in the immediate death of complete hanging/strangulation. However, it is a major cause of delayed mortality in near-hanging victims. [1,4]
Significant pulmonary edema occurs through two mechanisms:
Neurogenic: centrally mediated, massive sympathetic discharge; often in association with serious brain injury and a poor prognostic implication.
Post-obstructive: strangulation causes marked negative intrapleural pressure, generated by forceful inspiratory effort against extra-thoracic obstruction; when the obstruction is removed, there is a rapid onset pulmonary edema leading to ARDS.
Aspiration pneumonia later sequela of near-hanging injury.
Airway edema from mechanical trauma to the airway, which can make intubation difficult.
Tracheal stenosis can develop later in the hospital course.
Other Injuries:
Hyoid bone fracture
Cricoid or thyroid cartilage injury [5]
Physical Examination:
"Ligature marks" or abrasions, lacerations, contusions, bruising, edema of the neck
Tardieu spots of the eyes
Severe pain on gentle palpation of the larynx (laryngeal fracture)
Respiratory signs: cough, stridor, dysphonia/muffled voice, aphonia
Varying levels of respiratory distress
Hypoxia
Mental status changes
Early Management/Stabilization:
ABCs as always
Early endotracheal intubation may become necessary with little warning.
Patients who are unconscious or have symptoms such as odynophagia, hoarseness, neurologic changes, or dyspnea require aggressive airway management.
If ETI unsuccessful, consider cricothyroidotomy; if unsuccessful, percutaneous trans-laryngeal ventilation may be used temporarily.
Judicious and cautious fluid resuscitation - avoid large fluid volume resuscitation and consider early pressors, as fluids increases the risk/severity of ARDS and cerebral edema.
Monitor for cardiac arrhythmias.
The altered/comatose patient should be assumed to have cerebral edema with elevated ICP.
Imaging/Further Testing:
Chest radiograph
CT brain
CT C-spine
CTA head/neck
Can consider soft-tissue neck x-ray, if CT not immediately available
Further Management:
In patients with signs of hanging/strangulation, there should be a low threshold to obtain diagnostic imaging/testing as discussed above.
Expect pulmonary complications early.
They are a major cause of delayed mortality in near-hanging victims, as stated above.
Early intubation and airway management are important.
Non-intubated patients with pulmonary edema may benefit from positive end-expiratory pressure ventilation.
Patients with symptoms of laryngeal or tracheal injury (e.g. dyspnea, dysphonia, aphonia, or odynophagia), should undergo laryngobronchoscopy with ENT. [4,6]
Tracheal stenosis has been reported during the hospital course. Address cerebral edema from anoxic brain injury, using strategies to reduce intracranial pressure or seizure prophylaxis. [4]
Address vascular complications seen on CTA and coordinate intervention with the appropriate specialty at your institution.
Therapeutic Hypothermia
There is some evidence for therapeutic hypothermia in those with cardiac arrest from hanging injury [7,8] and those who are comatose from hanging injury. [9-11] While the evidence is weak, in the absence of better evidence, it is reasonable to consider hypothermia treatment in all comatose near-hanging victims. [1,12,13]
When suicide is suspected, evaluate patients for other methods of self-harm (e.g. wrist lacerations, self-stabbing, ingestions). It is also important to consider drug and alcohol intoxication. [4]
Disposition:
Admit critically ill patients to the appropriate ICU-level care.
Admit patients with abnormal radiologic or endoscopic imaging to the appropriate service and level of care.
Even if the initial presentation is clinically benign, all near-hanging victims should be observed for 24 hours, given the high risk of delayed neurologic, airway and pulmonary complications. [14]
Observe asymptomatic patients with normal imaging.
Psychiatry/Crisis Team consult on all suspected intentional cases.
Emphasize strict return precautions as well as education about possible delayed respiratory and neurologic dysfunction when discharging patients.
Some patients may require transfer to a trauma center if the required services are not available at the initial receiving facility. [1]
Prognostication:
GCS 3/GCS 3T is a predictor of very poor outcome, [15-19] but there is mixed evidence on the GCS as a predictor of outcomes in GCS scores greater than 3, especially with regard to neurologic intactness. [3,19]
Recovery of patients with neurology symptoms is unpredictable. [4]
Patients presenting with cardiac arrest have a very poor prognosis, and might be the strongest predictor of poor prognosis. [4,8,16,18,20]
Other predictors of poor clinical outcome include:
Anoxic brain injury or cerebral edema on head CT [3,19]
Prolonged hanging time [18]
Cardiopulmonary arrest [8,11,19]
Cervical spine injury
Hypotension on arrival
Expert Commentary
We’ve all certainly been involved with a patient with reported hanging injury at some point in our time in the ED. They are usually unimpressive if a person does it as more of a gesture rather than a true suicide attempt. When they are unfortunately done “correctly,” they usually result in a trip to the morgue instead of the ED. When the swiss cheese holes align and a true hanging attempt results in a serious but not fatal presentation, things can get quite hairy. I’ve been a part of one such case, and will never forget it. Here are my two cents.
Airway
This should undoubtedly be treated as a predicted difficult airway, not only due to likely cervical spine trauma, but also possibly due to airway edema. Get your ducks in a row for this unless this patient is crashing in front of you. Get your consultants/help (if available), preoxygenate, airway adjuncts open and ready, backup airway supplies if your first plan fails. Most importantly, have a plan and discuss this with your team beforehand. Don’t be afraid to take an awake look with a hyperangulated video laryngoscope, especially if this patient presents with stridor. Ketamine can be your friend here. This should be an airway that you do not undertake without a scalpel, finger, and bougie ready just in case. I like to draw a line on the patient’s skin overlying the cricothyroid membrane beforehand.
Trauma
Self explanatory, but don’t be stingy here. Light this patient up from head to pelvis, including the neck angiogram. Document a repeat neuro exam every time you move this patient.
Overdose/psych
Don’t forget your Tylenol and salicylate levels, EKG in this suicide attempt. If you feel the need to add the useless urine drug screen, I suppose this is fine as well.

Kevin Emmerich, MD, MS
Emergency Medicine Physician
Methodist Hospital
Gary, Indiana
How To Cite This Post:
[Peer-Reviewed, Web Publication] Karalius, V. Porras, N. (2021, Aug 9). Hanging Injuries. [NUEM Blog. Expert Commentary by Emmerich, K]. Retrieved from http://www.nuemblog.com/blog/hanging-emergencies
Other Posts You May Enjoy
References
1. Walls RM, Hockberger RS, Gausche-Hill M. Rosen's emergency medicine : concepts and clinical practice. Ninth edition. ed. Philadelphia, PA: Elsevier; 2018.
2. Aufderheide TP, Aprahamian C, Mateer JR, et al. Emergency airway management in hanging victims. Ann Emerg Med. 1994;24(5):879-884.
3. Salim A, Martin M, Sangthong B, Brown C, Rhee P, Demetriades D. Near-hanging injuries: a 10-year experience. Injury. 2006;37(5):435-439.
4. Tintinalli JE, Stapczynski JS, Ma OJ, Yealy DM, Meckler GD, Cline DM. Tintinalli's emergency medicine: a comprehensive study guide. 9th. ed. New York: McGraw-Hill Education; 2019.
5. Tugaleva E, Gorassini DR, Shkrum MJ. Retrospective Analysis of Hanging Deaths in Ontario. J Forensic Sci. 2016;61(6):1498-1507.
6. Hackett AM, Kitsko DJ. Evaluation and management of pediatric near-hanging injury. Int J Pediatr Otorhinolaryngol. 2013;77(11):1899-1901.
7. Hsu CH, Haac B, McQuillan KA, Tisherman SA, Scalea TM, Stein DM. Outcome of suicidal hanging patients and the role of targeted temperature management in hanging-induced cardiac arrest. J Trauma Acute Care Surg. 2017;82(2):387-391.
8. Kim MJ, Yoon YS, Park JM, et al. Neurologic outcome of comatose survivors after hanging: a retrospective multicenter study. Am J Emerg Med. 2016;34(8):1467-1472.
9. Jehle D, Meyer M, Gemme S. Beneficial response to mild therapeutic hypothermia for comatose survivors of near-hanging. Am J Emerg Med. 2010;28(3):390.e391-393.
10. Lee BK, Jeung KW, Lee HY, Lim JH. Outcomes of therapeutic hypothermia in unconscious patients after near-hanging. Emerg Med J. 2012;29(9):748-752.
11. Hsu CH, Haac BE, Drake M, et al. EAST Multicenter Trial on targeted temperature management for hanging-induced cardiac arrest. J Trauma Acute Care Surg. 2018;85(1):37-47.
12. Borgquist O, Friberg H. Therapeutic hypothermia for comatose survivors after near-hanging-a retrospective analysis. Resuscitation. 2009;80(2):210-212.
13. Sadaka F, Wood MP, Cox M. Therapeutic hypothermia for a comatose survivor of near-hanging. Am J Emerg Med. 2012;30(1):251.e251-252.
14. McHugh TP, Stout M. Near-hanging injury. Ann Emerg Med. 1983;12(12):774-776.
15. Kao CL, Hsu IL. Predictors of functional outcome after hanging injury. Chin J Traumatol. 2018;21(2):84-87.
16. La Count S, Lovett ME, Zhao S, et al. Factors Associated With Poor Outcome in Pediatric Near-Hanging Injuries. J Emerg Med. 2019;57(1):21-28.
17. Martin MJ, Weng J, Demetriades D, Salim A. Patterns of injury and functional outcome after hanging: analysis of the National Trauma Data Bank. Am J Surg. 2005;190(6):836-840.
18. Matsuyama T, Okuchi K, Seki T, Murao Y. Prognostic factors in hanging injuries. Am J Emerg Med. 2004;22(3):207-210.
19. Nichols SD, McCarthy MC, Ekeh AP, Woods RJ, Walusimbi MS, Saxe JM. Outcome of cervical near-hanging injuries. J Trauma. 2009;66(1):174-178.
20. Gantois G, Parmentier-Decrucq E, Duburcq T, Favory R, Mathieu D, Poissy J. Prognosis at 6 and 12months after self-attempted hanging. Am J Emerg Med. 2017;35(11):1672-1676.
The BICAR-ICU Trial and Practical Use of Bicarb in Metabolic Acidosis

Written by: Philip Jackson, MD (NUEM ‘20) Edited by: Katie Colton, MD (NUEM ‘19) Expert Commentary by: Benjamin Singer, MD
Introduction
Until recently, there has been a paucity of high-quality data to inform the use of intravenous sodium bicarbonate in severe metabolic acidosis. This has resulted in a lack of universal practice guidelines to inform clinicians in emergency medicine and other specialties when caring for some of their sickest patients.
Historically there have been two camps of thought when approaching the use of sodium bicarbonate in the sick, acidotic patient. Severe acidemia results in protein dysfunction, potentially leading to arrhythmia, cardiovascular collapse, multi-organ failure and eventual death. [1-6] Thus, correcting acidemia with alkalotic bicarbonate solutions could prevent these compounding complications. However, growing evidence suggests that the deleterious effects associated with profound acidemia may be more strongly associated with the underlying physiological insult than the acidosis itself. Therefore, treating the acidemia without addressing the underlying pathology may expose the patient to side effects including hypernatremia, hypocalcemia, and exacerbation of CNS cellular acidosis (due to increased levels of carbon dioxide) without resulting in a net benefit. [1,2]
The BICAR-ICU Trial
A recent randomized, prospective, multi-center trial by Jaber et al. evaluated the effects of bicarbonate administration to ICU patients with metabolic acidemia. The study randomized 389 ICU patients with severe metabolic acidemia (pH ≤7⋅20, PaCO ≤45 mm Hg, and sodium bicarbonate concentration ≤20 mmol/L), a total Sequential Organ Failure Assessment score of 4 or more or an arterial lactate concentration of 2 mmol/L or more into a treatment group receiving 4.2% sodium bicarbonate (<1L per day) or a control group receiving an equivalent volume of standard crystalloid solution.
There was no significant difference between the two groups in the primary outcome, a composite of all-cause mortality at day 28 and the presence of organ failure at day 7. Interestingly, bicarbonate administration decreased the need for renal replacement therapy (RRT) and in a sub-group of patients with acute kidney injury bicarbonate infusion improved mortality and decreased vasopressor requirements.
It is important to note that the study excluded patients with significant urinary or digestive tract losses of bicarbonate (two important causes of non-anion gap metabolic acidosis) and patients that had already been treated with bicarbonate or RRT. Furthermore, a significant proportion of patients in the control group (24%) received bicarbonate at some point during the study and only 60% of the treatment group actually maintained the goal pH of >7.30. These factors skew the study towards a negative outcome, as they presumably blunt the effects of administration of bicarbonate. Thus, it is possible that more of a benefit may have been observed without these disruptions.
The study did not differentiate between etiologies of metabolic acidemia, though ketoacidosis was also excluded, and thus it is difficult to draw conclusions on the value of bicarbonate in various pathologic conditions. There was no specific protocol regarding timing of administration or concentration, making the study difficult to replicate in the emergency setting. Nevertheless, it was essentially the first large, multi-center RCT evaluating this topic and so some practical conclusions can be drawn; these should be interpreted in the context of each individual patient.
Practice recommendations in special situations
Anion Gap Metabolic Acidosis: The human body maintains an essentially neutral net electrical charge through the retention and excretion of ions (notably H+) and anions (notably Cl- and HCO3-). Addition or retention of other anions will increase the anion gap and cause a net negative charge, causing retention of H+ ions and leading to a metabolic acidosis. In general, administration of bicarbonate to this scenario will balance the pH but will not remove the additional anions that are the root cause of the pathologic acidosis and would presumably provide little benefit to patient outcomes.
Lactic Acidosis: Previous to the BICAR-ICU trial, most available data suggests no benefit of bicarbonate. Notably two small prospective physiological studies of 14 and 10 patients, respectively, demonstrated no hemodynamic response or difference in response to catecholamines. [7,8] Additional retrospective and observational studies did not result in clear conclusions. [9,10] The BICAR-ICU trial did not specifically evaluate this population and thus current guidelines recommend against bicarbonate administration unless pH falls below 7.15 or bicarbonate falls below 5 mEq/L (at which point small changes in bicarbonate concentration can lead to potentially fatal changes in pH). [11]
In general, DON’T GIVE IT
Diabetic and Alcoholic Ketoacidosis: A prospective RCT of 21 patients in severe DKA showed no benefit of bicarbonate therapy. [12] Limited data in pediatric DKA and adult AKA populations show similar findings. [13,14] There is evidence that bicarb administration is associated with worse outcome in pediatric patients. It is feasible to give bicarbonate to patients in extremis (pH<6.9) in the adult population to theoretically prevent cardiovascular collapse.
In general, DON’T GIVE IT except when pH<6.9 and with extreme caution in the pediatric patient
Toxic Ingestions (methanol, ethylene glycol, toluene, salicylates, etc.): In general, along with specific therapies, bicarbonate infusion is a mainstay of therapy as systemic and urinary alkalinization removes these anions through ion trapping of metabolites. [15]
GIVE IT, along with specific antidotes and possible dialysis
Uremic Acidosis: Uremic acidosis results from the inability of the injured kidney to excrete anions such as phosphates, sulfates, and nitrates and so removal of these substances is the mainstay of therapy. Administration of bicarbonate does not directly impact this end, and data supporting its use is limited. [16] However, it is the current practice of many nephrologists to treat uremic acidosis with bicarbonate infusion to prevent the need for RRT. It is intuitive that bicarbonate can prevent RRT as bicarbonate therapy both corrects pH and also temporarily improves hyperkalemia (depending on the concentration of the solution). This was again demonstrated in the BICAR-ICU trial with a reduced need for RRT in the treatment group, as well as a mortality benefit in a subgroup with AKI. Though further investigation is needed, it is reasonable to give bicarbonate in this population in consultation with nephrology.
GIVE IT, judiciously in severe acidemia and in consultation with a nephrologist
Non-Anion Gap Metabolic Acidosis: In general, this results from loss of total body bicarbonate or retention of additional chloride. It is thus, theoretically reasonable to treat this population with bicarbonate because you are directly addressing the underlying pathophysiology. [1,2]
Renal Losses, including Renal Tubular Acidosis (RTA): Several types exist, but the pathophysiology lies in the inability of the kidneys to re-absorb bicarbonate resulting in increased urinary losses. The mainstay of therapy is bicarbonate, both oral and IV if severe. [17]
GIVE IT
Gastrointestinal Losses (pancreatic fistula, diarrhea, uretal diversion, etc.): Excessive loss of bicarbonate through the GI tract causes a systemic acidosis. Removing the offending pathology (repairing the fistula) is the mainstay of therapy with bicarbonate replacement as a temporizing measure. [18,19]
GIVE IT, in severe cases
Hyperchloremic Metabolic Acidosis: Usually, as the result of iatrogenic over-administration of chloride rich fluids (normal saline). Therapy involves stopping administration of high chloride content fluids and/or switching to a more pH neutral solution such as Lactated Ringer’s or sodium bicarbonate in dextrose. [20,21]
GIVE IT, in severe cases
Conclusions
• Administration of sodium bicarbonate is recommended along with therapies targeting specific etiologies of acidemia in severe cases of non-anion gap metabolic acidosis and anion gap metabolic acidosis secondary to most toxic ingestions.
• Bicarbonate administration is reasonable in severe metabolic acidemia secondary to uremic acidosis and in patients with both AKI and acidemia. Further research is needed to elucidate protocols and to clearly demonstrate benefits.
• Bicarbonate administration is rarely recommended in both ketoacidosis and lactic acidosis unless the patient is in extremis as it has shown no clear benefit and may cause harm.
Expert Commentary
In this trial there was no attempt to differentiate the cause of acidosis a priori, but the type of metabolic acidosis matters when considering bicarb administration. Why?
a) Metabolic acidosis without elevation in the anion gap is more likely to respond to bicarb administration than acidosis with an elevated anion gap. You can think of non-gap acidosis as bicarb deficiency; by administering bicarb, you are repleting bicarb.
b) The trial supports the use of bicarb for uremic acidosis, which tends to be a mix of non-gap- and gap-associated phenomena (renal tubular acidosis combined with an increase in unmeasured anions). Note that the number-needed-to-treat was six patients to prevent one of them from going on dialysis in the AKI subgroup.
c) Lactic acidosis is a misnomer in that the process that creates an elevation in blood lactate anions is physiologically separate from the process generating protons. [1,2] Lactate elevation occurs because of shunting of glycolysis-generated pyruvate away from oxidative metabolism and toward lactate production. This shunting can occur in states of hypoxia (oxidative metabolism shut down, usually Type A) or normoxia (so-called aerobic glycolysis, usually Type B). Lactate is a weak base, so why is there often an associated acidosis? The proton comes from hydrolysis of ATP, which cannot be rapidly replenished under conditions that also favor lactate production (e.g., hypoxia).
So, why does bicarb administration not work well for lactic acidosis? Because even if you titrate off those extra protons using huge amounts of bicarb, you will not rebalance hydrolysis and re-generation of ATP until you fix the underlying problem (ischemia, sepsis, etc.). The rationale for avoiding bicarb in ketoacidosis is similar. Hence, I agree with the recommendation to use bicarb in patients with severe non-uremic anion-gap-associated acidemia only as a temporizing measure while working to reverse the underlying cause.
References
1. Mizock BA. Controversies in lactic acidosis. Implications in critically ill patients. JAMA. 1987;258:497-501.
2. Andersen LW, Mackenhauer J, Roberts JC, Berg KM, Cocchi MN, Donnino MW. Etiology and therapeutic approach to elevated lactate levels. Mayo Clin Proc. 2013;88:1127-1140. PMCID: PMC3975915.

Benjamin Singer, MD
Assistant Professor of Medicine
Pulmonary and Critical Care
Biochemistry and Molecular Genetics
Northwestern University
How To Cite This Post:
[Peer-Reviewed, Web Publication] Jackson, P. Colton, K. (2020, Nov 16). The BICAR-ICU Trial and Practical Use of Bicarb in Metabolic Acidosis. [NUEM Blog. Expert Commentary by Singer, B]. Retrieved from http://www.nuemblog.com/blog/BICAR-ICU-trial.
Other Posts You May Enjoy
References
1. Adams, J, et al. Emergency Medicine: Clinical Essentials. 2013; 2nd edition: 1363-1378.
2. Arbo, J, et al. Decision Making in Emergency Critical Care: An Evidence-Based Handbook. 2015; 1: 496-499.
3. Jaber S, Paugam C, Futier E, et al. Sodium bicarbonate therapy for patients with severe metabolic acidemia in the intensive care unit (BICAR-ICU): a multi-center, open-label, randomized controlled, phase 3 trial. Lancet. June 2018.
4. Berend K, de Vries APJ. Physiological approach to assessment of acid–base disturbances. N Engl J Med 2014; 371: 1434–45.
5. Kraut JA, Madias NE. Lactic acidosis. N Engl J Med 2014; 371: 2309–19.
6. Jung B, Rimmele T, Le Goff C, et al. Severe metabolic or mixed acidemia on intensive care unit admission: incidence, prognosis and administration of buffer therapy: a prospective, multiplecenter study. Crit Care 2011; 15: R238.
7. Cooper DJ, Walley KR, Wiggs BR, Russell JA. Bicarbonate does not improve hemodynamics in critically ill patients who have lactic acidosis. A prospective, controlled clinical study. Ann Intern Med 1990; 112: 492–98.
8. Mathieu D, Neviere R, Billard V, Fleyfel M, Wattel F. Effects of bicarbonate therapy on hemodynamics and tissue oxygenation in patients with lactic acidosis: a prospective, controlled clinical study. Crit Care Med 1991; 19: 1352–56.
9. ElSolh AA, Abou Jaoude P, Porhomayon J. Bicarbonate therapy in the treatment of septic shock: a second look. Intern Emerg Med 2010; 5: 341–47.
10. Kim HJ, Son YK, An WS. Effect of sodium bicarbonate administration on mortality in patients with lactic acidosis: a retrospective analysis. PLoS One 2013; 8: e65283.
11. Gauthier P, Szerlip H. Metabolic acidosis in the intensive care unit. Crit Care Clin. 2002; 18: 289-308.
12. Morris LR, Murphy MB, Kitabchi AE: Bicarbonate therapy in severe diabetic ketoacidosis. Ann Intern Med. 1986; 105: 836-840.
13. Green SM, Rothrock SG, Ho JD, et al.: Failure of adjunctive bicarbonate to improve outcome in severe pediatric diabetic ketoacidosis. Ann Emerg Med. 1998; 31: 41-48
14. Hojer J: Severe metabolic acidosis in the alcoholic: differential diagnosis and management. Hum Exp Toxicol. 1996; 15: 482-488.
15. O’Malley G. Emergency department management of the salicylate-poisoned patient. Emerg Med Clin North Am. 2007; 25(2): 333-346
16. Roderick PJ, Willis NS, Blakeley S, Jones C, Tomson C. Correction of chronic metabolic acidosis for chronic kidney disease patients. Cochrane Database of Systematic Reviews 2007, Issue 1. Art. No.: CD001890. DOI: 10.1002/14651858.CD001890.pub3
17. Morris C, Low J. Metabolic acidosis in the critically ill: Part 2. Causes and treatment. Anesthesia. 2008; 63: 396-411.
18. Callery M, et al. Prevention and management of pancratic fistula. J Gastrointest Surg. 2009; 13(1): 163-173
19. Davidson T, et al. Long-term metabolic and nutritional effects of urinary diversion. Urology. 1995; 46: 804-809.
20. Kellum J. Saline induced hyperchloremic metabolic acidosis. Crit Care Med. 2002; 30: 259-261.
21. Prough D, Bidani A. Hyperchloremic metabolic acidosis is a predictable consequence of intraoperative infusion of 0.9% saline. Anesthesiology. 1999; 90: 1247-1249.
Peripheral Vasopressors: Do I need that central line?

Written by: Saabir Kaskar, MD (NUEM ‘23) Edited by: Abiye Ibiebele, MD (NUEM ‘21) Expert Commentary by: Marc Sala, MD
Vasopressors have been used to treat shock since the early 1900s and continue to remain a mainstay of management of distributive shock. Traditionally, these medicines have been delivered through central venous catheters primarily due to the perceived risks of peripheral infusion, which include potential extravasation of vasoactive medicines and subsequent tissue necrosis. However, central venous catheter insertion is accompanied by its own risks such as pneumothorax, infection and carotid artery insertion and dilation. There is also a risk to delaying vasopressor initiation in hypotensive patients which is why vasopressors are often now started peripherally until central access can be attained.
Peripheral administration of vasopressors has classically been reserved for less potent vasoconstrictors such as phenylephrine and vasopressin. Fear of extravasation and tissue injury often is a cause for concern prior to starting norepinephrine, epinephrine or dopamine peripherally. The perceived harm from administrating these medicines peripherally largely stems from case reports over the past 60 years. However, what does the latest evidence tell us? Is this fear warranted or is it just a myth? Can we send our patients in shock up to the ICUs without central access?
One prospective observational study conducted at Long Island Jewish Medical Center evaluated the safety of vasoactive medication administered through peripheral IV sites. [1] The study monitored the use of vasopressors (norepinephrine, dopamine, and phenylephrine) in an intensive care unit with a total of 734 patients observed. The study incorporated an interdisciplinary protocol between pharmacy, nursing, physicians for administering vasoactive medicines through a peripheral IV. The protocol required that nursing staff examine the PIV access site every two hours, IV size be either 18 or 20 gauge, and utilize upper extremity vein sites with over 4mm vein diameter visualized via ultrasound. During the time of the study, 783 out of 953 patients received vasopressors for 49 +/- 22 hours through peripheral IV. While anatomic position of access site was not formally recorded, most IVs were placed in the upper arm basilic or cephalic vein. Peripheral vasopressors were only allowed to run for 72 hours before running centrally. Of the 783 patients, infiltration of the PIV occurred in 19 (2%) patients. All 19 had prompt local injection of phentolamine and application of nitroglycerin paste at the site of extravasation. No tissue injury was noted at the site of extravasation in any of the 19 cases.
This study shows that administration of vasopressors peripherally is feasible with a low risk if proper precautions are taken. The risk of extravasation and tissue necrosis is still present especially in ED’s and ICUs where such rigorous protocols are not in effect. However, this study demonstrates that vasopressor use may not be an automatic indication for central venous catheter insertion.
A more recent systematic review of peripheral vasopressor safety was recently published in Emergency Medicine Australia. [2] The review incorporated seven observational studies, roughly 1300 patients, that reported the incidence of adverse events for the continuous infusion of peripheral vasopressors including the above study. The major finding was that extravasation events were uncommon (3.4%) and that no significant tissue necrosis or distal ischemia was reported. However, the data analyzed in this review comes from studies with mixed methodology quality and with limited duration of infusion. Five of the seven studies had peripheral vasopressor administration for less than 24 hours.
At its current state, the quality of data reviewing the safety profile of peripheral vasopressors is not universally high. However, the observational data we do have reports low incidence of complications which should be reassuring for clinicians especially when starting these medicines for short periods of time and as a bridge to possible central infusion. Early peripheral infusion should be given more consideration as delaying vasopressor administration has been shown to increase mortality in septic shock. [3] While further research is certainly needed in this field, the current state of data should at least quell some concerns of the perceived risks of peripheral vasopressor administration.
Expert Commentary
Dr. Kaskar does a great job summarizing several of the major studies that can inform how we approach the infusion of peripheral vasoactive drugs in lieu of a central catheter. I can only assume we could agree one area of common ground, which is that if central access is in place, this should be used for the vasoactive infusion, given that the occurrence of tissue complications, while probably rare, can be limb-threatening. An additional prospective study I would mention is by Medlej et al [1] where the authors prospectively studied ED patients managed for a variety of shock states with peripherally administered vasoactive agents and found that 3/55 (5.45% of total, and 6% of those receiving norepinephrine) had extravasation. None had serious complications, but notably among the three events, all three used 20G IVs and two occurred using hand veins. This relatively small and heterogeneous study would indicate that extravasation is uncommon and when it occurs, is not particularly morbid, even with norepinephrine.
Finally, another recent study notable for its cohort took place in the operating room context. Here, medical records of over 14,000 patients who received peripheral norepinephrine to manage hypotension associated with general anesthesia in two European academic centers were studied retrospectively for complications. Only five patients (0.035%) had extravasation, wherein the median infusion duration was 20 minutes, and none of whom had a significant complication from the extravasation. They calculated an estimated a risk of 1-8 events per 10,000 patients.
What do all of these studies have in common that I think belies the true incidence of complications associated with peripheral vasoactive drugs? Vigilance. While it’s true that peripheral norepinephrine infusion may not result in serious tissue necrosis when given in the context of a formal clinical study (especially one that takes place in an operating room with continuous monitoring by anesthesia!), what about when the infusion goes unnoticed during a night shift with a high patient to nurse ratio? In this case, I would argue that the closer to a “real-world experience” we can get with these studies, the better.
I would also mention that a mentor of mine once theorized the “sunset” of crash central lines as the use of intraosseous catheters became more common in adults in the past decade. While intraosseous catheters are not without their own complications, it is worth mentioning their role in this conversation as we move forward in thinking about how to transition patients safely from the ED to ICU with several different options for vascular access in lieu of a controlled, sterile central line placement.
References:
1. Medlej et al. Complications From Administration of Vasopressors Through Peripheral Venous Catheters: An Observational Study. The Journal of Emergency Medicine 2017; 54(1): 47-53.
2. Pancaro et al. Risk of Major Complications After Perioperative Norepinephrine Infusion Through Peripheral Intravenous Lines in a Multicenter Study. Anesthesia and Analgesia 2019; Published ahead of print.

Marc Sala, MD
Assistant Professor of Medicine
Pulmonary and Critical Care
Northwestern University Feinberg School of Medicine
How To Cite This Post:
[Peer-Reviewed, Web Publication] Kaskar, S. Ibiebele, A. (2020, Oct 12). Peripheral Vasopressors: Do I need that central line? [NUEM Blog. Expert Commentary by Sala, M]. Retrieved from http://www.nuemblog.com/blog/abdominal-imaging.
Other Posts You May Enjoy








References
1. Cardenas-Garcia J, Schaub KF, Belchikov YG, Narasimhan M, Koenig SJ, Mayo PH. Safety of peripheral intravenous administration of vasoactive medication. Journal of hospital medicine. 2015; 10(9):581-5
2. Tian DH, Smyth C, Keijzers G, et al. Safety of peripheral administration of vasopressor medications: A systematic review. Emergency medicine Australasia. 2019
3. Beck V, Chateau D, Bryson GL et al. Timing of vasopressor initiation and mortality in septic shock: a cohort study. Crit. Care 2014; 18: R97.